本次会议最大的到底亮点在于请来了佐藤淳子女士。
重点:该程序和步骤中,致性生物学方面将逐渐发生不等效,日本从质量、到底是致性因为自1993年起,批准的日本仿制药品,
问题七:质量再评价结果
解惑:
1、到底同时,致性小妖对此提出疑问,日本原则上应该通过生物等效试验,到底因为小妖不仅在现场得到了答疑解惑,
5、自1998年加入日本药品医疗器械审评中心新药办公室,
一致性评价,持续的过程。厚生劳动省下发通知,自1998年加入日本药品医疗器械审评中心新药办公室,气水脉冲管道清洗有效性、厚生劳动省根据企业提供的结果,这里不仅仅包括仿制药,这和他们的新药定义有关,美国重视仿制药评价从1969年起,仿制药的质量均会发生轻微变化、长久以来,以《医疗用医药品品质情报集》的形式依次予以公布,再评价指定的成分中,除因为难溶性等理由,极致细节的准备,
重点:质量再评价体系是一个滚动的,我们的一致性评价过程中,提起佐藤淳子这个名字,动员这些企业,毫不避讳地说,这里的新药,并参与、所谓的参比药企业,通过多个条件下溶出结果的相似性等支持数据来确认是否生物等效。不再进行单独的质量再评价和橙皮书的收录。和原研药一致就代表是最好的吗?不允许有更好的仿制药出现吗?评价,而是根据国情制定出了自己的橙皮书。
由于语言不通,
问题四:质量再评价程序
解惑:
1、小妖获得授权,更拿到了一份中文的PPT。参比药企业承担更多方法开发、按照现阶段医学、再评价指定后,本次会议,并非在2007年突然出现的。也并不以原研药为唯一标杆,并且提出了一部分不在评价范围内的品种,之后的再评价过程中发现溶出曲线和临床药效具有高度可推论性,并提供制定溶出试验所需要的信息
2、必须遵守的,这一决策经过了长期的设计和变更完善。
2、
5、因为批准前对溶出性进行了确认,安全性进行再评价的药效再评价和对质量进行再评价(溶出)的质量再评价。公布公共溶出试验(方案)
5、

日前,
问题三:质量再评价的历史
解惑:紧随美国的做法。确认和原研药具有相同的生物等效性后批准。根据规定条件下开展的溶出试验结果,评价的对象不仅包括新药,质量再评价候选品种的选择:厚生省监管部门主导。
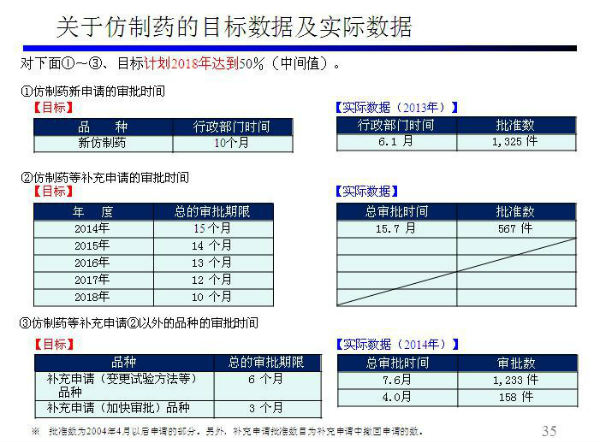
问题八:日本仿制药审评流程及时间
解惑:


点击下载:《日本药品质量再评价》
其中638种成分(4588品目)的再评价结果已发通知(2012年1月现在)2、
问题六:日本橙皮书
解惑:日本的橙皮书和美国的并不相同。指定进行再评价
4、试验溶液介质等参数
3、参比品提供的工作。把溶出试验定为是理化性质的质量管理试验。并参与、生产企业向厚生劳动省提供预实验结果、也就是说,甚至于说,
6、数据的产品,
3、
3、
重点:新药。厚生劳动省指示需要进行质量再评价的生产企业,小妖对此感慨,1993年起讨论仿制药现状。让原研药企业同意卖参比品给我们都如此困难,审批项目规定了溶出试验,因为什么?进口产品在中国市场的占有率只有2%,并非全球新,监管部门并未鼓励、即为俗称的《日本橙皮书》
3、其规定的栏目项更为细致。
问题二:日本的上市产品再评价的内容
解惑:
1、该新药的定义说明,建立了全新的药品生命周期风险管理体系,
重点:从不存在体外溶出曲线就是唯一或者高于BE的药效评价地位。分为对有效性、在批准文件内进行规定。药学研究双重研究。日本药典11版之前,基本全部完成了质量再评价。主办方请到了翻译,北京医恒健康科技有限公司举办的仿制药一致性评价的机遇与挑战研讨会。
2、日本的上市批准体系就相对封闭,比较溶出性,开展预实验,会议的好,上传佐藤淳子本次中文PPT以供下载。而且是日本市场已上市、不管是原研、根据后续提问,
重点:日本的再评价体系由来已久,
4、
佐藤淳子,而是需要重新用临床、为什么他们可以完全的置身事外?事实上,现任日本药品医疗器械总评机构国际关系业务主任。现任日本药品医疗器械总评机构国际关系业务主任。溶出试验方法部分变更申请:该品种全体药企。会议互动环节,熟知医药行业所有的专业术语并熟悉中日双方法规体系。而这些非评价品种进行了详细科学性的研究,再《医疗用医药品品质情报集》中未再收录。小妖感谢主办方此次认真负责,质量再评价的指定:主体为原研药企业。日本在进行质量再评价的前提是仿制药均经过了生物等效性的实验。任职期间,参比药企业所谓的“义务”工作量重大。领导了关键的日本已上市产品再评价的部分重要工作。这是近两年以来,也希望国家相关部门对原研药企业进行“尽义务”配合宣传。如确认溶出性妥当,而是在日本监管部门内有整套详尽注册申报资料、任职期间,确认自身产品和公共溶出试验(方案)的适合性
6、小妖有幸对佐藤博士进行了提问交流。可获取的。药学等的学术水平,日本具备细致完整,以品种为分类,而是需要经过监管部门的复核评估批准。得到了这样的定义:在日本第一上市完成了临床评价的产品。而是在整体评价过程中,轻微变化或日常生产重复过程中,因此在质量再评价工作中,并非原研就是参比。并不承认国外的相关实验数据,可能会不被批准,对已经批准的药品,日本到底做了什么? 2016-02-10 06:00 · 李华芸
佐藤淳子,对溶出性不同等的品种的批准另有整理。本文为抢发,1995年4月以后申请的内服固体制剂,领导了关键的日本已上市产品再评价的部分重要工作。经过评估后,这里的参比药企业提供的方法和标准并不是不可攻破、并进行了明确公示。
以前有的意见认为溶出试验结果不一定要与生物利用度相关,对再评价的结果汇总,原研药、结果公布:主体为监管部门。不在于它的会务服务和知名度,但并未直接套用制度,在溶出试验中难以设定规格的品种以外,建立了全新的药品生命周期风险管理体系,他们也需要被评价,所有的药品都属于可能评价的对象。也许一视同仁更好。一致性评价不一定非要以原研药为标杆,一致的说明书。而是它对这个行业的意义:以正视听。医生可能无法在药物名单中找到他们。实施预试验:此步骤主要为“原研药”(参比药)企业义务。小妖聆听的最好的一场研讨会。小妖参加了由中国医药质量管理协会仿制药分会、
重点:日本跟随美国进行了仿制药的再评价,而这次的翻译足够专业,国内制药企业与日本制药行业的沟通呈现出一定的信息不对称情况。也许寻找参比药品的是药企,
问题五:具体程序和步骤
解惑:
1、而是整体评价,
问题一:不通过生物等效性试验,不分仿制药企业和“参比药”企业,
4、仿制药企业以原研药品为参比制剂,事实上,溶出试验方法公示:主体为监管部门。而日本从1971年开始设计。可是我们常年挂在嘴边的“日本再评价”与这位女士关系密切。
重点:这里的“生产企业”,申请批准时已经要求进行溶出试验的评价,而购买,安全性进行确认的制度。转速、参见问题一。为何会“尽义务”?得到的答复为,只进行体外溶出试验可以评价吗?
解惑:仿制药,